Des chercheurs de l’école de médecine Icahn de Mount Sinai ont découvert que la plupart des mutations pathogènes présentent un faible risque de provoquer réellement la maladie. Crédit : avec l’aimable autorisation du laboratoire Do, Mount Sinai, N.Y., N.Y.
Les résultats d’une vaste étude de biobanque menée par des chercheurs de Mount Sinai pourraient aider les médecins à mieux évaluer le risque réel de maladie.
Imaginez que vous obteniez un résultat positif à un test génétique. Le médecin vous dit que vous avez une “variante génétique pathogène”, c’est-à-dire une séquence d’ADN connue pour augmenter les risques de contracter une maladie comme le cancer du sein ou le diabète. Mais quelles sont exactement ces chances – 10 % ? Cinquante pour cent ? Cent ? Actuellement, il n’est pas facile de répondre à cette question.
Pour répondre à ce besoin, des chercheurs de l’Icahn School of Medicine at Mount Sinai ont analysé les séquences d’ADN et les données des dossiers médicaux électroniques de milliers d’individus stockés dans deux grandes biobanques. Dans l’ensemble, ils ont découvert que le risque qu’une variante génétique pathogène provoque effectivement une maladie est relativement faible – environ 7 %. Néanmoins, ils ont également constaté que certaines variantes, telles que celles associées au cancer du sein, sont liées à un large éventail de risques de maladie. Les résultats, publiés dans JAMApourraient modifier la façon dont les risques associés à ces variantes sont signalés et, un jour, aider à guider la façon dont les médecins interprètent les résultats des tests génétiques.
“L’un des principaux objectifs de cette étude était de produire des statistiques utiles et avancées permettant d’évaluer quantitativement l’impact que les variantes génétiques connues causant des maladies peuvent avoir sur le risque de maladie d’un individu”, a déclaré Ron Do, PhD, professeur associé de génétique et de sciences génomiques et membre de l’Institut Charles Bronfman pour la médecine personnalisée à Icahn Mount Sinai.
Au cours des 20 dernières années, les scientifiques ont découvert des centaines de milliers de variantes susceptibles de provoquer diverses maladies. Cependant, en raison de la nature de ces découvertes, il a été difficile d’estimer – ou de fournir des statistiques sur – le risque réel que cela se produise pour chaque variante génétique. Jusqu’à présent, la plupart des estimations étaient fondées sur des études portant sur un petit nombre de sujets, qui faisaient partie d’une famille ayant des antécédents de maladie ou qui avaient été recrutés dans des cliniques spécialisées. Mais les études de ce type qui n’utilisent pas de grandes populations choisies au hasard peuvent produire des surestimations du risque posé par les variantes.
Dans cette étude, les chercheurs se sont attaqués au problème en recherchant 37 780 variantes connues dans les données de séquençage de l’ADN à grande échelle de 72 434 personnes, puis en analysant les dossiers médicaux de chaque individu pour trouver un diagnostic de maladie correspondant. La recherche approfondie a porté sur 29 039 participants au programme Bio de Mount Sinai.MoiBiobank de Mount Sinai et 43 395 participants qui faisaient partie de la UK Biobank.
L’étude a été dirigée par Iain S. Forrest, un candidat au doctorat en médecine dans le laboratoire du Dr Do, qui s’est inspiré d’une expérience clinique antérieure qu’il avait acquise dans le cadre d’une bourse post-baccalauréat aux National Institutes of Health (NIH).
“L’idée de l’étude est née d’une séance de brainstorming”, a déclaré M. Forrest. “Le Dr Do et moi-même avons discuté de la nécessité de disposer d’un meilleur système de classification des risques de maladie. À l’heure actuelle, les variantes sont classées selon des étiquettes générales telles que “pathogène” ou “bénigne”. Comme je l’ai appris en clinique, ces étiquettes comportent de nombreuses zones d’ombre. C’est alors que nous avons réalisé que les biobanques, qui relient les données de séquences d’ADN aux dossiers médicaux électroniques, constituent une opportunité inégalée pour répondre à ce besoin.”
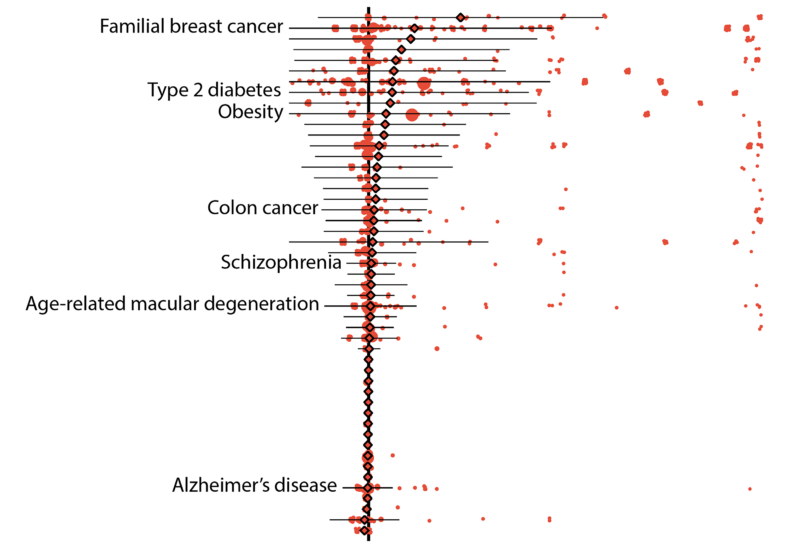
Les premiers résultats ont montré que 157 maladies de leur ensemble de données pouvaient être liées à 5 360 variants définis comme “pathogènes” par ClinVar, une bibliothèque publique largement référencée et soutenue par le NIH, ou comme “perte de fonction” telle que prédite par des algorithmes bioinformatiques. En moyenne, la “pénétrance”, ou la probabilité qu’une variante soit liée au diagnostic d’une maladie, était faible, à savoir 6,9 %. De même, la différence de risque moyenne, qui décrit l’augmentation du risque de maladie pour un individu qui possède la variante par rapport à un individu qui ne la possède pas, était également faible.
“Au début, j’ai été assez surpris par les résultats. Les risques que nous avons découverts étaient plus faibles que ce à quoi je m’attendais”, a déclaré le Dr Do. “Ces résultats soulèvent des questions sur la façon dont nous devrions classer les risques de ces variantes.”
Malgré ces résultats, les risques associés à certains variants génétiques sont restés élevés. Par exemple, les variants pathogènes des gènes du cancer du sein. BRCA1 et BRCA2 ont toutes deux une pénétrance moyenne de 38 %, les variantes individuelles se situant entre zéro et 100 %.pour cent.
D’autres résultats ont démontré d’autres avantages de l’utilisation des données des biobanques. Dans un exemple, les chercheurs ont pu calculer les risques de variantes individuelles qui sont associées à des troubles liés à l’âge, comme certaines formes de diabète de type 2 et les cancers du sein et de la prostate. En moyenne, la pénétrance de ces variantes était d’environ 10 % pour les personnes de plus de 70 ans, alors qu’elle était d’environ 8 % pour celles de plus de 20 ans.
L’équipe a également constaté que la présence de certains variants pouvait dépendre de l’origine ethnique de l’individu et a identifié plus de 100 variants que l’on trouve spécifiquement chez les personnes d’origine non européenne.
Enfin, les auteurs ont énuméré plusieurs façons dont l’étude elle-même aurait pu sous-estimer ou surestimer les risques signalés.
“Bien que d’autres recherches soient nécessaires, nous pensons que cette étude est un bon premier pas vers la fourniture éventuelle aux médecins et aux patients des informations précises et nuancées dont ils ont besoin pour établir des diagnostics plus précis”, a déclaré le Dr Do.
Référence : “Population-Based Penetrance of Deleterious Clinical Variants” par Iain S. Forrest, BS ; Kumardeep Chaudhary, PhD ; Ha My T. Vy, PhD ; Ben O. Petrazzini, BS ; Shantanu Bafna, MS ; Daniel M. Jordan, PhD ; Ghislain Rocheleau, PhD ; Ruth J. F. Loos, PhD ; Girish N. Nadkarni, MD ; Judy H. Cho, MD et Ron Do, PhD, 25 janvier 2022, .
DOI : 10.1001/jama.2021.23686
Ce travail a été soutenu par les National Institutes of Health (GM124836, GM007280, HL139865, et HL155915).