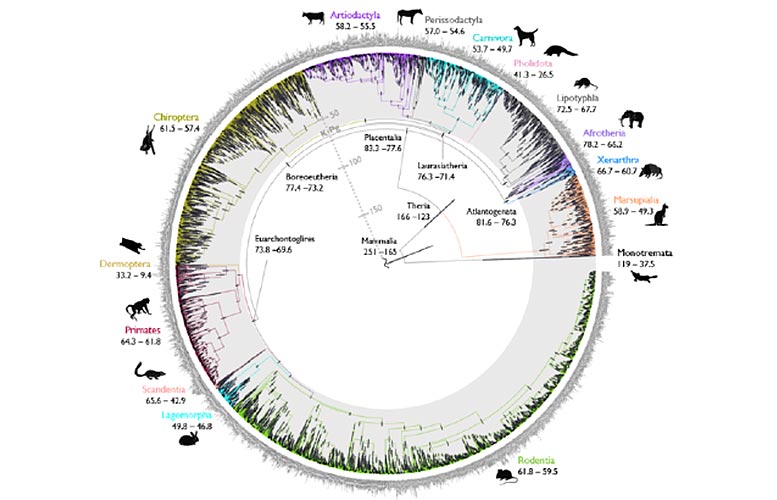
Arbre de vie des mammifères. Crédit : Mario dos Reis Barros et Sandra Alvarez-Carretero
Une nouvelle étude, publiée le 22 décembre 2021, dans la revue La nature, a fourni la chronologie la plus détaillée de l’évolution des mammifères à ce jour.
La recherche décrit une nouvelle approche informatique rapide pour obtenir des arbres évolutifs datés avec précision, appelés « arbres temporels ». Les auteurs ont utilisé la nouvelle méthode pour analyser un ensemble de données génomiques de mammifères et répondre à une question de longue date quant à savoir si les groupes de mammifères placentaires modernes sont apparus avant ou après le Crétacé-Extinction de masse paléogène (K-Pg), qui a anéanti plus de 70 pour cent de toutes les espèces, y compris tous les dinosaures.
Les découvertes confirment les ancêtres des groupes de mammifères placentaires modernes après l’extinction de K-Pg qui s’est produite il y a 66 millions d’années, réglant une controverse sur les origines des mammifères modernes. Les mammifères placentaires constituent le groupe de mammifères vivants le plus diversifié et comprennent des groupes tels que les primates, les rongeurs, les cétacés, les carnivores, les chiroptères (chauves-souris) ainsi que les humains.
L’équipe de recherche était dirigée par le Dr Mario dos Reis (Queen Mary University of London) et le professeur Phil Donoghue (Université de Bristol), et comprenait des scientifiques de Queen Mary, Université de Bristol, UCL, collège impérial de Londres, et l’Université de Cambridge.
Le Dr Sandra Álvarez-Carretero, auteur principal de l’article de l’UCL (alors à Queen Mary), déclare : « En intégrant des génomes complets dans l’analyse et les informations fossiles nécessaires, nous avons pu réduire les incertitudes et obtenir une chronologie évolutive précise. Les groupes de mammifères modernes ont-ils coexisté avec les dinosaures, ou sont-ils apparus après l’extinction de masse ? Nous avons maintenant une réponse définitive.
« La chronologie de l’évolution des mammifères est peut-être l’un des sujets les plus controversés de la biologie évolutive. Les premières études ont fourni des estimations d’origine pour les groupes placentaires modernes au plus profond du Crétacé, à l’époque des dinosaures. Au cours des deux dernières décennies, les études ont oscillé entre les scénarios de diversification post-K-Pg et pré-K-Pg. Notre calendrier précis règle le problème », ajoute le professeur Donoghue, co-auteur principal de l’article.
Approche rapide pour l’analyse du génome
Avec des projets de séquençage mondiaux produisant désormais des centaines à des milliers de séquences de génomes, et avec des plans imminents de séquençage de plus d’un million d’espèces, les biologistes évolutionnistes auront bientôt une mine d’informations à leur disposition. Cependant, les méthodes actuelles pour analyser les vastes ensembles de données génomiques disponibles et créer des chronologies évolutives sont inefficaces et coûteuses en calculs.
« Induire des chronologies évolutives est un objectif fondamental de la biologie. Cependant, les méthodes de pointe reposent sur l’utilisation d’ordinateurs pour simuler des chronologies évolutives et évaluer les plus plausibles. Dans notre cas, cela a été difficile en raison du gigantesque ensemble de données analysé, impliquant des données génétiques de près de 5 000 espèces de mammifères et 72 génomes complets », explique le Dr dos Reis.
Dans cette étude, les chercheurs ont développé une nouvelle approche bayésienne rapide pour analyser un grand nombre de séquences génomiques, tout en tenant compte des incertitudes dans les données. « Nous avons résolu les obstacles informatiques en divisant l’analyse en sous-étapes : d’abord en simulant des chronologies à l’aide des 72 génomes, puis en utilisant les résultats pour guider les simulations sur les espèces restantes. L’utilisation de génomes réduit l’incertitude car elle permet de rejeter les chronologies non plausibles des simulations », explique le Dr dos Reis.
« Notre pipeline de traitement de données a fourni autant de données génomiques que possible pour autant d’espèces de mammifères. C’était difficile car les bases de données génétiques contiennent des inexactitudes et nous avons dû développer une stratégie pour identifier les échantillons de mauvaise qualité ou les données mal étiquetées qui ont dû être supprimées », ajoute le Dr Asif Tamuri, co-auteur principal de l’article de l’UCL, qui était responsable de l’assemblage de l’ensemble de données génomiques des mammifères.
Plus efficace et durable
Grâce à leur nouvelle approche, l’équipe a pu réduire le temps de calcul de cette analyse complexe de plusieurs décennies à plusieurs mois. « Si nous avions essayé d’analyser cet ensemble de données sur les grands mammifères dans un superordinateur sans utiliser la méthode bayésienne que nous avons développée, nous aurions dû attendre des décennies pour déduire l’arbre temporel des mammifères. Imaginez combien de temps cette analyse pourrait prendre si nous utilisions nos propres PC », explique le Dr Álvarez-Carretero. « De plus, nous avons réussi à réduire le temps de calcul d’un facteur 100. Cette nouvelle approche permet non seulement l’analyse de jeux de données génomiques, mais aussi, en étant plus efficace, réduit considérablement le CO2 émissions dues à l’informatique », poursuit le Dr Álvarez-Carretero.
La méthode développée dans l’étude pourrait être utilisée pour s’attaquer à d’autres chronologies évolutives controversées qui nécessitent l’analyse de grands ensembles de données. En intégrant la nouvelle approche bayésienne aux futurs génomes des projets Darwin Tree of Life et Earth BioGenome, l’idée d’estimer une échelle de temps évolutive fiable pour l’Arbre de Vie semble désormais à portée de main.
Référence : « Une chronologie au niveau des espèces de l’évolution des mammifères intégrant des données phylogénomiques » par Sandra Álvarez-Carretero, Asif U. Tamuri, Matteo Battini, Fabrícia F. Nascimento, Emily Carlisle, Robert J. Asher, Ziheng Yang, Philip CJ Donoghue et Mario dos Reis, le 22 décembre 2021, La nature.
DOI : 10.1038 / s41586-021-04341-1


