Par
Cellules de mammifères génétiquement modifiées avec succès à l’aide de la méthode STAMPScreen. Crédit : Institut Wyss de l’Université Harvard
Le pipeline STAMPScreen aide à rationaliser les études génétiques dans les cellules de mammifères
Les ingénieurs généticiens d’aujourd’hui ont une pléthore de ressources à leur disposition : un nombre toujours croissant d’ensembles de données massifs disponibles en ligne, des outils d’édition de gènes très précis comme CRISPR et des méthodes de séquençage de gènes bon marché. Mais la prolifération des nouvelles technologies ne s’est pas accompagnée d’une feuille de route claire pour aider les chercheurs à déterminer quels gènes cibler, quels outils utiliser et comment interpréter leurs résultats. Ainsi, une équipe de scientifiques et d’ingénieurs du Wyss Institute for Biologically Inspired Engineering de Harvard, de la Harvard Medical School (HMS) et du AVEC Media Lab a décidé d’en faire un.
L’équipe Wyss a créé un pipeline intégré pour effectuer des études de dépistage génétique, englobant chaque étape du processus, de l’identification des gènes cibles d’intérêt au clonage et au dépistage rapide et efficace. Le protocole, appelé Sequencing-based Target Assurement and Modular Perturbation Screening (STAMPScreen), est décrit dans Méthodes de rapports de cellule, et les algorithmes open source associés sont disponibles sur GitHub.
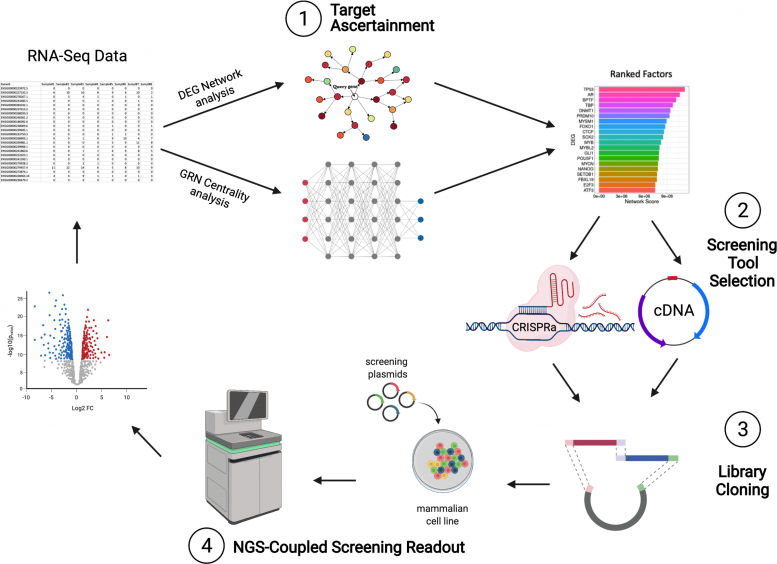
Le flux de travail STAMPScreen est un pipeline intégré qui permet aux chercheurs d’analyser rapidement et facilement une base de données expérimentale pour les gènes d’intérêt potentiels (1), de choisir l’outil de dépistage à utiliser (2), de créer une bibliothèque de dépistage (3) et d’utiliser la prochaine génération séquençage pour cribler des gènes in vivo (4). Les étapes individuelles peuvent également être utilisées dans d’autres workflows. Crédit : Institut Wyss de l’Université Harvard
« STAMPScreen est un flux de travail rationalisé qui permet aux chercheurs d’identifier facilement les gènes d’intérêt et d’effectuer des criblages génétiques sans avoir à deviner quel outil utiliser ou quelles expériences effectuer pour obtenir les résultats souhaités », a déclaré l’auteur correspondant Pranam Chatterjee, Ph. D., un ancien étudiant diplômé du MIT Media Lab qui est maintenant le chercheur Carlos M. Varsavsky au HMS et au Wyss Institute. « Il est entièrement compatible avec de nombreuses bases de données et systèmes existants, et nous espérons que de nombreux scientifiques pourront tirer parti de STAMPScreen pour gagner du temps et améliorer la qualité de leurs résultats. »
La frustration est la mère de l’invention
Chatterjee et Christian Kramme, co-premier auteur de l’article, étaient frustrés. Les deux scientifiques essayaient d’explorer les fondements génétiques de différents aspects de la biologie – comme la fertilité, le vieillissement et l’immunité – en combinant les forces des méthodes numériques (pensez aux algorithmes) et du génie génétique (pensez au séquençage des gènes). Mais ils ont continué à rencontrer des problèmes avec les divers outils et protocoles qu’ils utilisaient, qui sont monnaie courante dans les laboratoires scientifiques.
Les algorithmes qui prétendaient passer au crible les gènes d’un organisme pour identifier ceux qui ont un impact significatif sur un processus biologique donné pourraient dire quand le modèle d’expression d’un gène a changé, mais n’ont fourni aucun aperçu de la cause de ce changement. Lorsqu’ils ont voulu tester une liste de gènes candidats dans des cellules vivantes, il n’était pas immédiatement clair quel type d’expérience ils devaient mener. Et bon nombre des outils disponibles pour insérer des gènes dans les cellules et les cribler étaient coûteux, longs et rigides.

Co-premier auteur de l’article, Christian Kramme, sur son banc à l’Institut Wyss. Crédit : Institut Wyss de l’Université Harvard
« J’utilisais des méthodes connues sous le nom de Golden Gate et Gateway pour cloner des gènes dans des vecteurs pour des expériences de criblage, et il m’a fallu des mois et des milliers de dollars pour cloner 50 gènes. Et en utilisant Gateway, je ne pouvais pas physiquement coder les gènes pour identifier celui qui pénétrait dans quel vecteur, ce qui était une exigence cruciale pour ma conception expérimentale basée sur le séquençage en aval. Nous avons pensé qu’il devait y avoir une meilleure façon de faire ce genre de recherche, et quand nous ne pouvions pas en trouver une, nous avons relevé le défi de la créer nous-mêmes », a déclaré Kramme, qui est un étudiant diplômé du Wyss Institute et du HMS. ,
Kramme s’est associé au co-premier auteur et membre du laboratoire de l’Église, Alexandru Plesa, qui éprouvait des frustrations identiques en créant des vecteurs génétiques pour son projet. Kramme, Plesa et Chatterjee se sont ensuite mis au travail pour définir ce qui serait nécessaire pour créer une plate-forme de bout en bout pour le dépistage génétique qui fonctionnerait pour tous leurs projets, allant de l’ingénierie des protéines à la fertilité et au vieillissement.
Des morceaux au banc
Pour améliorer la première étape de la recherche génétique – identifier les gènes d’intérêt à étudier – l’équipe a créé deux nouveaux algorithmes pour aider à répondre au besoin d’outils informatiques capables d’analyser et d’extraire des informations à partir des ensembles de données de plus en plus volumineux qui sont générés. passant par séquençage de nouvelle génération (NGS). Le premier algorithme prend les données standard sur le niveau d’expression d’un gène et les combine avec des informations sur l’état de la cellule, ainsi que des informations sur les protéines connues pour interagir avec le gène. L’algorithme attribue un score élevé aux gènes fortement connectés à d’autres gènes et dont l’activité est corrélée à de grands changements au niveau cellulaire. Le deuxième algorithme fournit des informations de plus haut niveau en générant des réseaux pour représenter les changements dynamiques de l’expression des gènes au cours de la différenciation des types cellulaires, puis en appliquant des mesures de centralité, telles que l’algorithme PageRank de Google, pour classer les principaux régulateurs du processus.
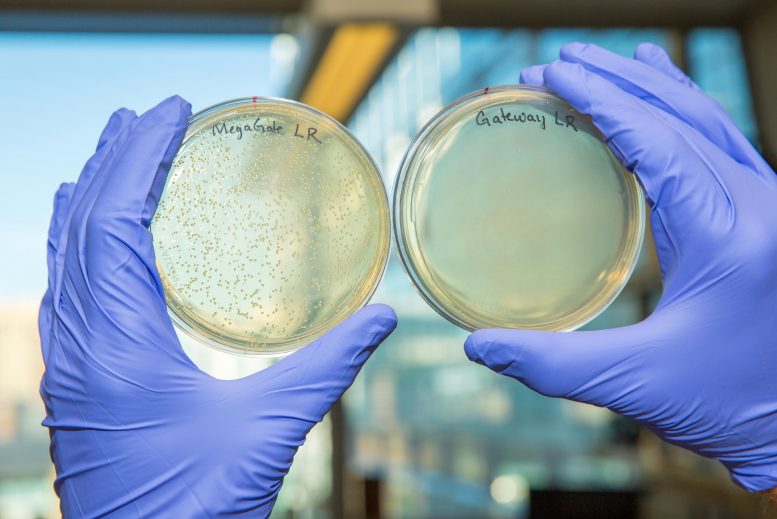
MegaGate, une nouvelle méthode de clonage de gènes cibles d’intérêt dans des vecteurs, est beaucoup plus efficace pour produire des vecteurs porteurs de gènes réussis (à gauche) que d’autres méthodes existantes comme Gateway (à droite). Crédit : Institut Wyss de l’Université Harvard
« La partie informatique des études génétiques est comme un jeu Jenga : si chaque bloc de la tour représente un gène, nous recherchons les gènes qui constituent la base de la tour Jenga, ceux qui soutiennent le tout. La plupart des algorithmes ne peuvent que vous dire quels gènes sont dans la même rangée, mais les nôtres vous permettent de déterminer à quelle distance ils se trouvent en haut ou en bas de la tour, afin que vous puissiez rapidement identifier ceux qui ont la plus grande influence sur la cellule. État en question », a déclaré Chatterjee.
Une fois les gènes cibles identifiés, le protocole STAMPScreen passe de l’ordinateur portable au laboratoire, où des expériences sont effectuées pour perturber ces gènes dans les cellules et voir quel effet cette perturbation a sur la cellule. L’équipe de chercheurs a systématiquement évalué plusieurs outils de perturbation génétique, y compris des outils complémentaires ADN (ADNc) et plusieurs versions de CRISPR dans les cellules souches pluripotentes induites par l’homme (hiPSC), les premières comparaisons directes connues réalisées entièrement dans ce type de cellule très polyvalent mais difficile.
Ils ont ensuite créé un nouvel outil qui permet d’utiliser CRISPR et l’ADNc dans la même cellule pour débloquer des synergies entre les deux méthodes. Par exemple, CRISPR peut être utilisé pour désactiver l’expression de toutes les isoformes d’un gène, et l’ADNc peut être utilisé pour exprimer séquentiellement chaque isoforme individuellement, permettant des études génétiques plus nuancées et réduisant considérablement l’expression de fond des gènes hors cible.
Numérisation des codes-barres de la bibliothèque
La prochaine étape de nombreuses expériences génétiques consiste à générer une bibliothèque de criblage pour introduire des gènes dans les cellules et observer leurs effets. En règle générale, les fragments de gènes sont insérés dans des plasmides bactériens (morceaux d’ADN circulaires) à l’aide de méthodes qui fonctionnent bien pour les petits morceaux d’ADN, mais sont difficiles à utiliser lors de l’insertion de gènes plus gros. De nombreuses méthodes existantes reposent également sur une technique appelée Gateway, qui utilise un processus appelé recombinaison du phage lambda et la production d’une toxine pour tuer toute bactérie qui n’a pas reçu de plasmide avec le gène d’intérêt. La toxine contenue dans ces plasmides est souvent difficile à manipuler en laboratoire et peut être inactivée par inadvertance lorsqu’une séquence de « code-barres » est ajoutée à un vecteur pour aider les chercheurs à identifier le plasmide porteur de gènes que le vecteur a reçu.
Kramme et Plesa travaillaient avec Gateway lorsqu’ils ont réalisé que ces problèmes pouvaient être résolus s’ils éliminaient la toxine et la remplaçaient par de courtes séquences sur le plasmide qui seraient reconnues et coupées par un type d’enzyme appelé méganucléases. Les séquences de reconnaissance des méganucléases n’apparaissent dans les gènes d’aucun organisme connu, garantissant ainsi que l’enzyme ne coupera pas accidentellement le gène inséré lui-même pendant le clonage. Ces séquences de reconnaissance sont naturellement perdues lorsqu’un plasmide reçoit un gène d’intérêt, rendant ces plasmides immunisés contre la méganucléase. Cependant, tous les plasmides qui ne reçoivent pas avec succès le gène d’intérêt conservent ces séquences de reconnaissance et sont coupés en morceaux lorsqu’une méganucléase est ajoutée, ne laissant qu’un pool pur de plasmides contenant le gène inséré. La nouvelle méthode, que les chercheurs ont surnommée MegaGate, a eu un taux de réussite de clonage de 99,8 % et leur a également permis de coder facilement leurs vecteurs.
« MegaGate résout non seulement bon nombre des problèmes que nous avons rencontrés avec les anciennes méthodes de clonage, mais il est également compatible avec de nombreuses bibliothèques de gènes existantes telles que TFome et hORFeome. Vous pouvez essentiellement prendre Gateway et les méganucléases du commerce, les assembler avec une bibliothèque de gènes et une bibliothèque de vecteurs de destination à code-barres, et deux heures plus tard, vous avez vos gènes d’intérêt à code-barres. Nous avons cloné près de 1 500 gènes avec, et nous n’avons pas encore eu d’échec », a déclaré Plesa, qui est un étudiant diplômé du Wyss Institute et du HMS.
Enfin, les chercheurs ont démontré que leurs vecteurs à code-barres pouvaient être insérés avec succès dans des hiPSC vivants et que des pools de cellules pouvaient être analysés à l’aide de NGS pour déterminer quels gènes délivrés étaient exprimés par le pool. Ils ont également utilisé avec succès diverses méthodes, notamment ARN-Seq, TAR-Seq et Barcode-Seq, pour lire à la fois les codes-barres génétiques et l’intégralité des transcriptomes des hiPSC, permettant aux chercheurs d’utiliser l’outil qu’ils connaissent le mieux.
L’équipe prévoit que STAMPScreen pourrait s’avérer utile pour une grande variété d’études, y compris des études de réseaux de régulation des voies et des gènes, le criblage de facteurs de différenciation, les caractérisations de médicaments et de voies complexes et la modélisation de mutations. STAMPScreen est également modulaire, permettant aux scientifiques d’en intégrer différentes parties dans leurs propres flux de travail.
« Il existe un trésor d’informations hébergées dans des ensembles de données génétiques accessibles au public, mais ces informations ne seront comprises que si nous utilisons les bons outils et méthodes pour les analyser. STAMPScreen aidera les chercheurs à accéder plus rapidement aux moments eurêka et à accélérer le rythme de l’innovation en génie génétique », a déclaré l’auteur principal George Church, Ph.D., membre de la faculté Wyss Core qui est également professeur de génétique à HMS et professeur de santé. Sciences et technologie à Harvard et au MIT.
« Au Wyss Institute, nous visons des solutions « moonshot » percutantes aux problèmes urgents, mais nous savons que pour aller sur la lune, nous devons d’abord construire une fusée. Ce projet est un excellent exemple de la façon dont notre communauté innove à la volée pour permettre des percées scientifiques qui changeront le monde pour le mieux », a déclaré le directeur fondateur de Wyss, Don Ingber, MD, Ph.D., qui est également le Judah Folkman Professeur de biologie vasculaire au HMS et au programme de biologie vasculaire du Boston Children’s Hospital, ainsi que professeur de bio-ingénierie à la Harvard John A. Paulson School of Engineering and Applied Sciences.
Référence : « An Integrated Pipeline for Mammalian Genetic Screening » 27 septembre 2021, Méthodes de rapports de cellule.
Les autres auteurs de l’article incluent Helen Wang, Bennett Wolf, Merrick Smela, Xiaoge Guo, Ph.D., et Richie Kohman, Ph.D. du Wyss Institute et du HMS.


